A Escolha de Sofia – A judicialização de medicamento não-incorporado e o custo-efetividade no SUS

1. A discussão sobre a concessão judicial de medicamento não incorporado à política pública de saúde tem suscitado grandes debates. O Supremo Tribunal Federal, ao julgar os Temas 6 e 1234, que culminaram com as súmulas vinculantes 61 e 60, respectivamente, deu um largo e positivo passo para auxiliar no debate, criando critérios que auxiliam na questão da chamada judicialização da saúde.
Embora haja progresso, ainda há muitos espaços de discussões.
As novas tecnologias são introduzidas no mercado em uma velocidade que frequentemente impede sua avaliação segundo os elevados padrões de Medicina Baseada em Evidências preconizados nos temas e pela comunidade científica. Sem falar em incorporação de medicamentos com estudos apenas de fase II
É crucial distinguir o papel das agências de registro e autorização de venda, como o Food and Drug Administration (FDA) dos Estados Unidos e a Agência Europeia de Medicamentos (EMA) e a própria Agência de Vigilância Sanitária – ANVISA, da tarefa desempenhada por órgãos ou instituições responsáveis pela incorporação da tecnologia em políticas públicas, como a nossa CONITEC, ou congêneres internacionais como o NICE, CADTH, do Reino Unido e Canadá, respectivamente, para ficar em apenas dois exemplos.
Sequer a diferenciação entre o papel da ANVISA e da CONITEC, por vezes, é bem compreendida. E a confusão entre a função dos diferentes entes estrangeiros é ainda maior, sendo recorrente o argumento de aprovação pelo FDA para justificar a concessão judicial, o que não corresponde ao critério fixado pelos temas.
Contudo, mesmo em cenários de estudos de fase III que demonstrem segurança, eficácia e efetividade, o alto custo de determinados fármacos representa uma barreira, conforme a exigência legal de demonstração de custo-efetividade para a incorporação ao SUS (art. 19-Q, da Lei nº 8080/90).
2. Estudos de fase II, para fármacos destinados a fatores específicos como sobrevivência, casos graves de saúde sem que haja tecnologia disponível ou doenças progressivas, têm sido objeto de aprovação pelo FDA na forma de fast track, um procedimento simplificado cuja autorização é concedida em até 60 dias
Este ponto parece-me essencial, porque em ambas as hipóteses os estudos ainda são bastante precários, embora haja uma delimitação quanto à população beneficiada pela tecnologia e a demonstração da indispensabilidade desta para o cuidado da saúde, ante a inexistência de substituto terapêutico.
Ocorre que este tipo de medicação invariavelmente está associada a elevados custos. Descabe aqui discutir os motivos pelos quais alguns medicamentos têm valores milionários, embora isso seja objeto de discussão internacional.
O fenômeno não é exclusivo da aprovação de registro no modo fast track, havendo medicamentos de alto custo registrados após a conclusão de estudos de fase III, mediante procedimento regular que, igualmente, tem elevados valores fixados junto à CMED, após o seu registro.
E aqui surge a raiz do problema que se busca examinar: a existência de medicamentos devidamente registrados na ANVISA, muitas vezes medicamentos órfãos e/ou para doenças raras, cuja precificação é milionária.
A existência de evidência científica de alto nível é um dos requisitos da incorporação de tecnologia em saúde. A inexistência de substituto terapêutico reforça essa recomendação, tanto que autoriza até mesmo seu registro mediante fast track.
Contudo, a precificação acaba por afastar o segundo requisito legal, que é a existência de custo-efetividade.
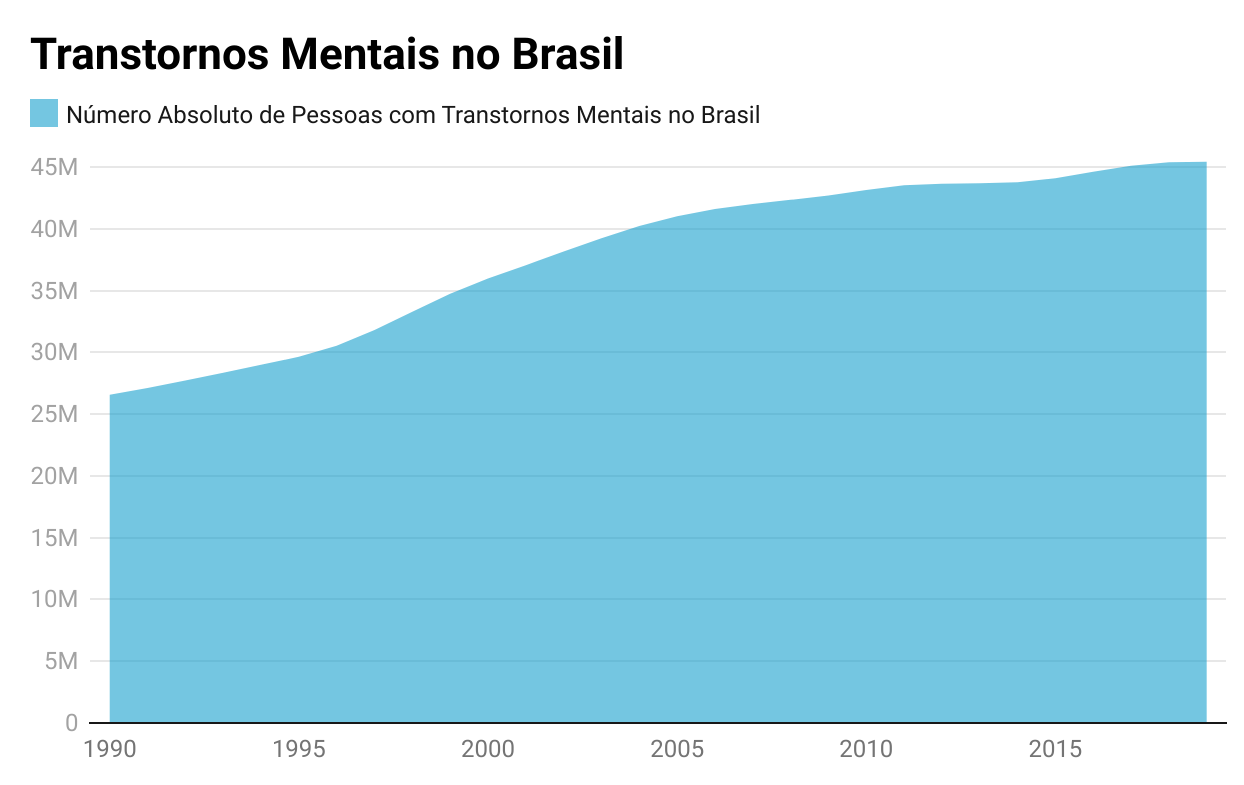
E são muitos os medicamentos que se acham nessa situação e impactam sobremaneira a judicialização da saúde e os orçamentos públicos. São fármacos que trazem esperanças reais aos pacientes, com elevados níveis de evidências que, em tese, recomendam seu fornecimento pelo sistema público de saúde.
Os benefícios esperados não se resumem à melhora do paciente, mas também impactam sua qualidade de vida, a vida de seus familiares e, muitas vezes, desoneram o próprio sistema de saúde em decorrência de despesas inerentes à morbidade, às comorbidades ou às consequências destas.
A judicialização desses medicamentos revela aspectos bastante incômodos, porque há estudos apontando a efetividade, com registro na Anvisa e aprovação pelo FDA e EMA.
Algumas tecnologias buscam aumentar em alguns meses a sobrevida de pacientes, ou mesmo diminuir um pouco a progressão de graves enfermidades que acometem pacientes, o que já é bastante positivo. Mas há medicamentos cujo tratamento permite a cura ou, pelo menos, tornar a vida da pessoa muito mais próxima daquela de outras pessoas que não têm a doença.
3. Nos casos em que há existência de evidência científica robusta, o cerne do debate reside na análise do custo-efetividade do tratamento
A complexidade transcende a análise da lide individual, pois a decisão judicial impacta a política pública de saúde. A questão jurídica não pode ser havida numa perspectiva isolada, porque o direito que assistirá ao demandante é idêntico ao direito de muitos outros que, acometidos da mesma condição de saúde, embora não tenham judicializado.
Considere-se uma doença rara aquela com incidência de 1 pessoa para cada 25.000 crianças nascidas. Com natalidade brasileira aproximada de 2,52 milhões por ano
Se o custo desse tratamento for estimado em R$ 1 milhão de reais, por ano, para cuidar de todas as crianças com esta doença rara, o custo seria de R$ 100 milhões de reais por ano. Se o tratamento for de uso prolongado, há que se multiplicar esse custo pelo tempo necessário ao tratamento (por exemplo 10 anos). Essa multiplicação pelo número de anos implica que todos aqueles que estivessem nessa faixa, igualmente deveriam se beneficiar da tecnologia. Assim, se fornecido para todos os 100 pacientes necessitados, haveria um custo anual de R$ 100 milhões. Multiplicada por todas as faixas etárias, considerando apenas 10 anos de uso, exigiria um orçamento de R$ 1 bilhão por ano.
As projeções orçamentárias preliminares demonstram o desafio da administração pública na alocação de recursos, evidenciando o dilema do custo de oportunidade.
Das precárias contas percebe-se a dificuldade que a administração pública terá para optar entre comprar este medicamento ou outros remédios ou, ainda, investir em outra política pública de saúde.
Não se pode ignorar os aspectos positivos que o investimento em saúde trará para o paciente, a família e até mesmo para a sociedade, porque uma pessoa saudável representa não apenas um ser humano e todas as suas potencialidades, mas também um impacto menor por outras demandas sanitárias.
Todavia, administrar é fazer escolhas. E essas competem precipuamente aos gestores. Ao Poder Judiciário incumbe a correção de ilegalidades e a exigência do cumprimento de políticas públicas, mas não a substituição do gestor na esfera de suas escolhas discricionária.
As comparações entre despesas e custeio têm sido objeto de demonstração pela administração
4. Fazer escolhas é complexo, difícil e, certamente, doloroso. Mas é exatamente disto que se trata administrar: estabelecer as melhores políticas públicas, mediante debate plural e democrático, considerando as variáveis e recursos existentes.
A administração pública deve pautar suas decisões por critérios técnico-científicos, sendo os órgãos especializados os responsáveis por indicar segurança, eficácia, efetividade e custo-efetividade (art. 19-Q, § 2º, da Lei nº 8080/90).
A questão é sobre política pública, devendo a administração mover-se segundo os marcos legais existentes. E o foro apropriado para debate não é o Poder Judiciário. A este cabe, apenas, analisar se a administração pública atua segundo os marcos legais. Para analisar os aspectos técnico-científicos, há um órgão competente no seio do Ministério da Saúde: a CONITEC.
O Ministério da Saúde, por intermédio da CONITEC, deve ser provocado para iniciar um processo de Avaliação de Tecnologia em Saúde (ATS), com prazo de 180 (cento e oitenta) dias, prorrogável por mais 90 (noventa), que inclui consulta e audiência pública, garantindo debate plural e democrático (art. 19-R, da Lei nº 8080/90), seguindo um rito (§ 1º, do artigo referido) que prevê consulta pública e audiência pública. A participação da sociedade e de especialistas é fundamental para a transparência e aderência social às decisões da CONITEC.
Certamente o resultado deste procedimento não tem a velocidade de uma liminar judicial, mas representa uma solução consistente, sólida e equânime para todos que necessitam da tecnologia. E esta decisão da CONITEC, como toda medida administrativa, é possível de ser confrontada pela via judicial, desde que a discussão não verse sobre o mérito do ato administrativo, mas apenas sobre seus eventuais vícios.
Os limiares de custo-efetividade e/ou custo de oportunidade
Há uma grande distância entre o valor praticado pela biofarmacêutica e os limiares de custo-efetividade (art. 19-Q, § 3º, da Lei do SUS) fixados pela legislação
Como destacado pelo Ministério da Saúde, por ocasião da divulgação dos limiares, Diversos países já estabeleceram limiares de custo-efetividade, como Austrália, Canadá e Chile. A Colômbia recentemente produziu artigo científico em que trata do tema e propôs para o limiar um valor de 1 PIB per capita. Já o National Institute for Health and Care Excellence (NICE), do Reino Unido e referência internacional, estabeleceu o limiar há 29 anos, decisão realizada por um comitê constituído pela legislação do país
No Brasil, recomendou-se a adoção de aproximadamente um PIB per capita ao ano, por QALY, valor esse flexibilizado em situações como em doenças pediátricas, com redução importante de sobrevida ajustada pela qualidade, em doenças raras e na atenção às pessoas em vulnerabilidade social, sendo três vezes o valor padrão do limiar proposto.
As soluções administrativas merecem deferência judicial
Porém, na via judicial, o mais adequado modo para discussão sobre incorporação de novas políticas públicas de saúde será por meio de ações coletivas. Estes medicamentos órfãos e voltados para portadores de doenças raras são um bom exemplo de casos que deveriam ser objeto de proteção por meio de tutelas coletivas.
A judicialização individual para concessão de medicamentos não incorporados na política pública de saúde gera contribuição marginal para a melhoria estrutural do SUS e resulta na alocação forçada de recursos orçamentários. Se é certo que essas ações fizeram melhorar o sistema de incorporação, com a criação da CONITEC e previsão de um procedimento próprio de avaliação de tecnologia (a partir da Lei nº 12.401/2011, que alterou a Lei nº 8080/90), não é menos certo que muitos recursos orçamentários
A via da ação coletiva estruturante, com todas as dificuldades que lhe são inerentes, é o instrumento adequado para garantir isonomia e o dimensionamento do problema e produzir uma solução sistêmica. O julgamento do tema 1234 é um bom exemplo.
Os dramas aflitivos das ações individuais, colocando o julgador em autênticas “Escolha de Sofia
5. Ações estruturantes colocam em foco não apenas o problema, mas também os principais atores dessa discussão, com os entes estatais responsáveis pela incorporação e fornecimento dos medicamentos, pacientes e/ou associação de seus representantes, bem como a indústria farmacêutica.
Com efeito, não basta colocar o Estado e os pacientes em campos opostos. A litigância há que envolver um terceiro componente, que é a indústria farmacêutica.
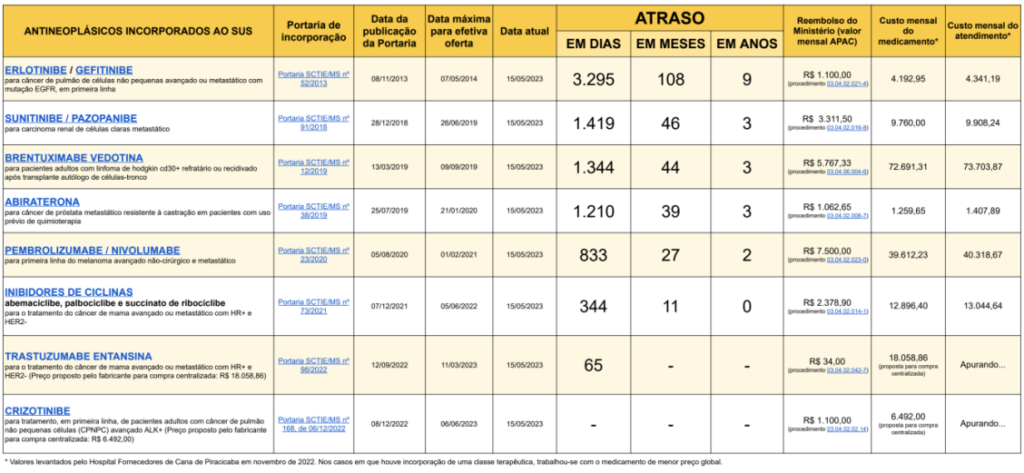
O laboratório que produz um medicamento que custa milhões de reais ao ano não é apenas parte do problema. Ele tem que ser parte da sua solução. E, neste campo da precificação, ainda estamos engatinhando na busca de soluções, às custas de milhões ou bilhões de reais já gastos com tecnologias milionárias (no ano de 2025 foram cerca de R$ 2 bilhões de reais aplicados em decorrência de ordens judiciais)
A discussão sobre custos não é um fenômeno exclusivo do Brasil, como informa o Telessaúde
Equivale dizer, o mundo inteiro está preocupado com os valores, mas no Brasil parece haver maior dificuldade em compreender que saúde tem custo, bem como que os recursos são finitos.
E a via da judicialização individual tem propiciado que a indústria farmacêutica promova o registro do produto na Anvisa, passando o medicamento a ser reivindicado judicialmente com custeio pelo Sistema Único de Saúde, sem sequer passar pelo crivo adequado da Conitec e do Ministério da Saúde.
Antes mesmo que qualquer outro país do mundo (muitos deles com desenvolvimento econômico muito superior ao do Brasil) tenha incorporado a tecnologia, ela passa a ser custeada pelo orçamento público por imposição judicial. Isso num sistema que compreende mais de 210 milhões de potenciais usuários.
Não se quer demonizar a indústria, até porque é ela quem traz as novas tecnologias para um maior cuidado das pessoas. Mas o tema da precificação de medicamentos preocupa o mundo, como destacado pela OMS
O Poder Judiciário brasileiro não pode fechar os olhos a essa realidade.
Não raras vezes fármacos são incorporados em outros sistemas públicos de saúde, por preços com cláusulas de confidencialidade ou descontos substanciais, como aponta outra Nota Técnica do telessaúde
E, no Brasil, divulga-se a incorporação em sistemas de saúde estrangeiros, fomentando a concessão judicial, mesmo com preços astronômicos que foram fixados por ocasião do registro na ANVISA.
6. Em suma:
6.1. A incorporação de tecnologia em saúde exige a interação de múltiplos atores, sendo o debate plural indispensável para que os escassos recursos em saúde sejam bem utilizados.
6.2. A relação de custo-efetividade é um requisito legal essencial para a incorporação tecnológica (Art. 19-Q, Lei nº 8.080/90), sendo vedado ao Poder Judiciário determinar o fornecimento de tecnologia que não atenda a esse pressuposto.
6.3. A intervenção judicial deve conferir deferência às decisões administrativas, seja pela legitimidade destas, seja pela expertise técnica empregada nas soluções.
6.4. Tratando-se de ação que busca a concessão judicial de procedimento ou medicamento não incorporado, a ação coletiva estruturante é a via apropriada, de modo que a tutela judicial seja extensível a todos que estejam na mesma situação fático-jurídica, bem como dimensionar o problema e trazer, para a solução, todos os atores.
6.5. O Brasil necessita de nova política pública de precificação dos medicamentos de alto custo, de modo a garantir acesso à população às novas tecnologias, com a incorporação dos fármacos no Sistema Único de Saúde a preço justo.
6.6. A “Escolha de Sofia” colocada ao Poder Judiciário nas ações individuais falseia o problema, tanto na perspectiva da ausência do consequencialismo, quanto na outorga individual de algo que deveria ser coletivo.
Imagem foto de holigil 27 na Unsplash.