Preços de medicamentos no Brasil: uma tragédia evitável

1 Introdução
Pouco se fala sobre as determinantes comerciais da saúde (Commercial Determinants of Health – CDOH). De uma maneira bastante simples e resumida, elas consistem nos fatores, sistemas ou práticas comerciais que interferem na saúde da população, tanto para o bem (determinantes comerciais positivas) como para o mal (determinantes comerciais negativas).
O que se propõe a discutir neste momento é uma das principais determinantes comerciais negativas da saúde pública, que consiste no preço dos medicamentos.
Segundo estimativa feita na Assembleia Mundial da Saúde da OMS em 2019, as despesas com medicamentos levam aproximadamente cem milhões de pessoas à pobreza por ano
Conforme bem observado no Health and Human Rights Resource Guide, “agora, mais do que nunca, os elevados preços dos medicamentos essenciais são cada vez mais entendidos como um problema global que afeta todos os países, e não apenas os em desenvolvimento” (livre tradução dos autores).
É usual que os consumidores obtenham, nas farmácias, elevados descontos nas compras de medicamentos. Ainda que, no âmbito individual, uma pessoa saia de uma drogaria muito feliz ao comprar seu remédio com um grande desconto ou com o chamado “convênio com o fabricante”, isso não deveria ser motivo para regozijo, mas uma lástima.
Na verdade, quando se adquire o medicamento com desconto, possivelmente o valor inicial da oferta era muito superior ao devido. Essa tragédia se agrava em relação aos menos favorecidos e nas localidades onde não há concorrência entre os varejistas.
Por outro lado, os tratamentos para doenças raras e ultrarraras – nova tendência da indústria farmacêutica – têm possibilitado a oferta de medicamentos com preços milionários, sem que se possa entender as razões para a fixação dos valores de venda ou mesmo conhecer os preços praticados em outros países, se eventualmente lá disponíveis.
Para ficar em três exemplos, medicamentos com terapia genética para tratar Atrofia Muscular Espinhal – AME (Zolgensma), outro para Hemofilia B (Hemgenix) e o recente remédio para cuidados de Distrofia Muscular de Duchene – DMD (Elevidys), foram precificados por US$ 2,1 milhões, US$ 3,5 milhões e US$ 3,2 milhões, respectivamente.
São muitas as questões que o tema suscita acerca da regulação e dos encaminhamentos possíveis, elencando-se resumidamente alguns tópicos relevantes.
2 Transparência no investimento para pesquisa e desenvolvimento e nos seus resultados
O investimento da indústria em pesquisa e desenvolvimento (P&D) de novas tecnologias em saúde representa menos de 50% de seu custo, segundo dados do Washington Post. Márcia Angell
Aliás, a falta de transparência em relação ao investimento em P&D, aos gastos com publicidade dos medicamentos e aos incentivos à sua prescrição
Reportagem publicada no site do jornal “O Globo” noticiou que, de acordo com dados compilados pela Bloomberg, “o faturamento da farmacêutica dinamarquesa Novo Nordisk com o Ozempic e o Wegovy (dois medicamentos cuja eficácia para o tratamento de obesidade e comorbidades a ela relacionadas, como diabetes, riscos cardíacos e pressão alta foi recentemente comprovada) é tão alto que as vendas dos dois medicamentos logo ultrapassarão todo o orçamento de pesquisa da empresa nas últimas três décadas”.
O sigilo muitas vezes atinge inclusive os preços de comercialização dos medicamentos, pactuados em acordos de negociação com países que os adquirem para os respectivos sistemas públicos de saúde. Isso compromete a análise para precificação por parte de agências reguladoras de outros países, que não sabem sequer quais os valores praticados internacionalmente.
3 Critérios de precificação e reajustamento dos medicamentos
Além da necessária transparência, é indispensável que sejam repensados os critérios de precificação, revisão ou atualização das tabelas CMED, que sofrem reajuste anual segundo padrões inflacionários a despeito do preço de vários medicamentos diminuírem ao longo do tempo. A tabela CMED é elaborada tomando como base preços de registro em nove países. No entanto, nem sempre os valores tabelados no estrangeiro são aqueles efetivamente praticados.
A desconexão entre os valores das tabelas da CMED e a realidade do mercado pode ser facilmente aferida, como citado anteriormente, nos inúmeros casos em que as farmácias concedem grandes descontos nas vendas de medicamentos aos consumidores, bastando que, para tanto, o comprador cadastre o seu CPF no estabelecimento. Isso demonstra que para atuarem com uma margem de lucro viável economicamente, as empresas podem vender os medicamentos bem abaixo dos preços máximos estabelecidos.
Há várias situações em que o valor de venda direta ao consumidor efetivamente praticado é inclusive igual ou inferior ao Preço Máximo de Venda ao Governo (PMVG) estabelecido pela CMED, que corresponde ao preço de fábrica sobre o qual ainda incide o redutor do Coeficiente de Adequação de Preço (CAP).
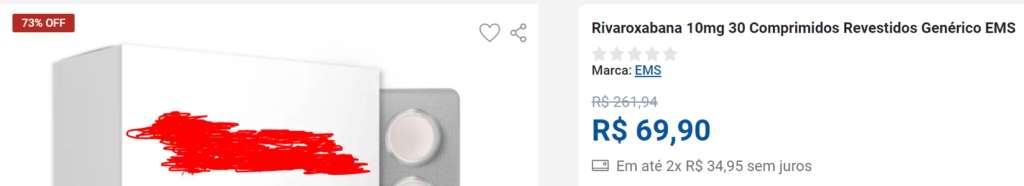
Tome-se como exemplo a rivaroxabana, medicamento utilizado para o tratamento de trombose e embolia pulmonar. Na apresentação de 10mg, uma caixa com 30 comprimidos do laboratório EMS S/A tem o PMVG fixado em R$ 158,22 pela CMED (tabela vigente em setembro de 2024), considerada a alíquota máxima do ICMS (22%).
Em rápida consulta a sites de vendas na internet, pode-se encontrar o mesmo medicamento, na mesma apresentação, com preço ao consumidor de R$ 69,90, em que o vendedor oferece um desconto de 73% sobre o preço original:
Outra farmácia anuncia o mesmo produto pelo valor de R$ 60,99, com desconto de 75%:
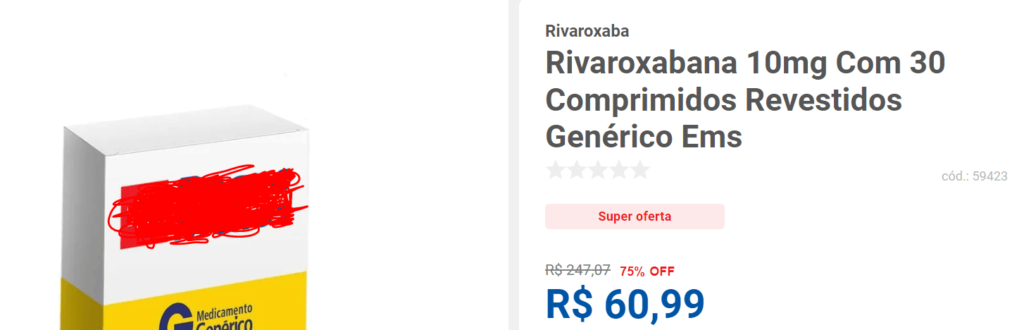
O preço final praticado por algumas farmácias quando vendem ao consumidor é inferior à metade do preço máximo de venda do medicamento ao Poder Público, e tudo isso dentro da sistemática permitida pelo tabelamento existente.
A discrepância entre a tabela CMED e a realidade do mercado, ao menos no que diz respeito a medicamentos já difundidos e que contam com alguma concorrência, é de clareza solar, fazendo com que, na prática, a estipulação de preços máximos não produza qualquer efeito mercadológico. No mundo real, só o poder público, os consumidores que adquirem os fármacos em localidades onde não haja concorrência e aqueles hipossuficientes é que acabam, na maioria das vezes, pagando os preços mais elevados.
4 Momento da fixação do preço dos medicamentos
Outro ponto relevante no tocante à precificação dos medicamentos é o momento da fixação inicial do seu preço. Para que um determinado produto seja vendido no país é necessário o prévio registro na Agência Nacional de Vigilância Sanitária (ANVISA), na forma da Lei nº 6.360/76, ocasião em que a empresa indicará o preço que pretende praticar (art. 16, VII), embora a própria lei autorize a dispensa de informações econômicas em casos específicos (art. 16, § 2º).
Todavia, depois de autorizado o registro é que se inicia o efetivo processo de precificação, com a inclusão do valor na Tabela CMED mediante procedimento previsto na Lei nº 10.742/2003, regulado pela Resolução CMED nº 2/2004. A CMED possui prazo que varia entre 60 a 90 dias para estabelecer o preço, após o que será autorizada a comercialização segundo o processo solicitado pelo fabricante.
A autorização do registro sem a prévia precificação representa um vácuo prejudicial ao consumidor, que ficará sujeito ao preço livremente pretendido pelo fabricante pelo menos até que a CMED estabeleça o justo valor. Assim, é indispensável que o registro de um medicamento na ANVISA seja precedido de precificação ou que ambos os atos ocorram concomitantemente.
Esse hiato entre o registro de um medicamento e a fixação de seu preço de tabela pode ser bastante longo. Isso impede que as novas tecnologias registradas pela ANVISA sejam avaliadas para possível disponibilização pelo SUS, já que a CONITEC deve analisar, quando recebe os pedidos de incorporação, também a relação de custo x efetividade do tratamento dentro dos limiares estabelecidos, além do impacto orçamentário que referida incorporação traria (art. 19-Q, §2º, II, e §3º, da Lei nº 8.080/90). Sem que se saiba o preço de um medicamento, é certo que essa verificação – e, consequentemente, eventual incorporação – ficam prejudicadas.
No julgamento do Tema 1234 de Repercussão Geral (RE 1.366.243), em que o Supremo Tribunal Federal tratou de uma série de questões relacionadas à judicialização da saúde no Brasil, foi constituída uma comissão especial composta por representantes dos entes federativos e das mais diversas instituições envolvidas na judicialização com o objetivo de buscar algum consenso em relação às medidas a serem adotadas. Dentre as propostas feitas pela comissão, algumas se relacionam justamente à questão agora abordada.
O ideal seria que a precificação pudesse ser contemporânea ao registro, sendo decorrência lógica do seu pedido, a ser instruído com todos os elementos necessários para que a CMED, no bojo do próprio procedimento assim instaurado, já estabelecesse os preços máximos a serem observados nas vendas.
5 As repercussões da precificação na incorporação dos tratamentos ao SUS
A incorporação de novas tecnologias de saúde ao SUS depende da análise, pela CONITEC, não apenas das evidências científicas de segurança, eficácia e efetividade do tratamento, mas também de questões econômicas ligadas à relação de custo x efetividade e ao impacto orçamentário de eventual disponibilização pelo sistema público de saúde. O mesmo ocorre em relação à avaliação de incorporação feita pela ANS em relação à saúde suplementar.
Especificamente no que diz respeito à relação de custo x efetividade, a CONITEC, em atenção ao comando do art. 19-Q, § 3º, da Lei nº 8.080/90, já emitiu suas recomendações sobre “O Uso de Limiares de Custo-Efetividade nas decisões em Saúde”. A comissão fixou limiares dentro dos quais, segundo a sua avaliação técnica, os custos com a incorporação de novas tecnologias de saúde ao SUS são vantajosos quando comparados aos resultados esperados com o tratamento e acima dos quais, consequentemente, os gastos não compensariam o benefício à saúde esperado.
Para possibilitar uma avaliação tecnicamente qualificada de todos esses aspectos de ordem econômica pelas agências responsáveis, é indispensável que o arcabouço regulatório da precificação dos medicamentos seja moderno, confiável e eficiente nos propósitos a que se destina, especialmente o de fazer com que os preços praticados atendam não apenas aos interesses da indústria farmacêutica, mas também dos destinatários de seus produtos.
6 Compartilhamento de riscos com a indústria
Além das questões relacionadas à precificação dos medicamentos, existem outras medidas que podem servir como determinantes comerciais positivas no objetivo de tornar os tratamentos médicos economicamente acessíveis aos cidadãos.
Uma delas é a celebração de acordo de compartilhamento de riscos entre o SUS – ou mesmo entre operadoras de planos de saúde – e os fabricantes de medicamentos.
Muitos dos novos medicamentos que chegam ao mercado com promessas revolucionárias – e, consequentemente, com valores altíssimos – não contam com todas as evidências científicas relacionadas aos desfechos clínicos relevantes para um sistema de saúde satisfatoriamente demonstradas, especialmente quando comparadas a outros tratamentos já disponíveis no mercado.
Em outras situações, os desfechos clínicos já observados ainda não permitem aferir com precisão uma relação de custo x efetividade ou de custo x oportunidade favorável à sua disponibilização pelo SUS ou por operadoras de planos de saúde. Neste cenário de incerteza, é razoável que os riscos de um possível não atendimento das expectativas do Poder Público com a disponibilização do tratamento à população necessitada sejam compartilhados com o fabricante ao invés de assumidos em sua integralidade pelo erário.
Os acordos de compartilhamento de riscos, em que pese já venham sendo utilizados largamente em outros países, ainda são pensados de forma tímida e com contornos não muito claros no nosso país. Na experiência brasileira, pode ser mencionada a Portaria GM nº 1.297/2019, do Ministério da Saúde, que instituiu projeto piloto de acordo de compartilhamento de risco para acesso ao medicamento Spinraza (Nusinersena) para o tratamento da Atrofia Muscular Espinhal (AME 5q) tipos II e III no âmbito do Sistema Único de Saúde – SUS.
Não houve, entretanto, notícias recentes sobre o êxito do procedimento, tampouco a extensão deste tipo de acordos para outras hipóteses de medicamentos ultracaros. Enfim, o regime experimental de compartilhamento de risco teve seu mérito de introduzir esse novo modo de aquisição e fornecimento de tecnologia nova para as pessoas que dela necessitem, mas há críticas ponderáveis ao modelo, como bem apontado por Ramos, Thomazi e Duarte Júnior
7 Revisão da estratégia de investimento em pesquisa e desenvolvimento
O modelo atual de pesquisa e desenvolvimento de medicamentos é concentrado fortemente na iniciativa privada.
A busca do lucro, por si, não é algo ruim. Pelo contrário, é ela que incentiva a iniciativa privada a trabalhar para o desenvolvimento científico e para descobrir tratamentos de saúde cada vez mais eficazes e que revertem em proveito da sociedade. A atividade econômica relacionada ao desenvolvimento de novas tecnologias em saúde deve, portanto, ser incentivada.
Novas tecnologias são pensadas e desenvolvidas primordialmente de acordo com o proveito econômico que elas proporcionam para as empresas e não com as necessidades de saúde pública. O resultado disso é a existência de uma vasta gama de doenças negligenciadas e de tratamentos inovadores, mas economicamente inacessíveis à população, além da falta de equidade de acesso entre sociedades de alta e baixa renda.
Por essas razões, é fundamental que o Poder Público atue na pesquisa e desenvolvimento de tecnologias em saúde, seja diretamente, com pesquisas custeadas e promovidas em instituições públicas, seja indiretamente, incentivando a iniciativa privada a operar de acordo com o interesse público.
A Constituição atribuiu expressamente ao SUS a responsabilidade por “participar da produção de medicamentos, equipamentos, imunobiológicos, hemoderivados e outros insumos” (art. 200, I) e por “incrementar, em sua área de atuação, o desenvolvimento científico e tecnológico e a inovação” (art. 200, V). Além disso, determinou que “a pesquisa científica básica e tecnológica receberá tratamento prioritário do Estado, tendo em vista o bem público e o progresso da ciência, tecnologia e inovação” (art. 218, § 1º).
Recentemente foi publicada a Lei nº 14.977, de 18/09/2024, que, acrescentando o art. 19-W à Lei nº 8.080/90, determinou que laboratórios farmacêuticos públicos produzam os princípios ativos destinados ao tratamento das doenças determinadas socialmente e que, para isso, o poder público fica autorizado a financiar, estimular, promover e buscar parcerias com outros laboratórios para transferência de tecnologia.
8 Patentes Farmacêuticas
Os direitos sobre a propriedade intelectual, mais especificamente as patentes de medicamentos novos e inovadores, também devem ser objeto de especial atenção na busca pela acessibilidade dos preços dos fármacos.
Não se discute que uma proteção adequada aos direitos de propriedade intelectual é indispensável para incentivar o investimento privado na pesquisa e desenvolvimento científicos na saúde. Por outro lado, o acesso às novas tecnologias é uma questão de saúde pública e de proteção a direito fundamental que não pode ficar refém de regras de mercado e dos interesses de empresas privadas.
No âmbito internacional, as patentes são objeto de regulamentação pelo Acordo Sobre Aspectos dos Direitos de Propriedade Intelectual Relacionados ao Comércio (Acordo TRIPS). Sobre ele, a Organização Mundial do Comércio assim se manifestou por meio da Declaração de Doha, ocasião em que deixou clara a necessidade da conjugação das patentes farmacêuticas com a proteção da saúde pública:
4. Nós concordamos que o Acordo TRIPS não impede e não deveria impedir seus membros de adotar medidas para proteger a saúde pública. Em conseqüência, enquanto reiteramos nosso compromisso com o Acordo TRIPS, nós afirmamos que o acordo pode e deve ser interpretado e implementado de modo a apoiar o direito dos membros da OMC de proteger a saúde pública e, em particular, de promover o acesso aos medicamentos para todos.
Assim sendo, nós reafirmamos o direito dos membros da OMC de utilizarem, em toda sua extensão, as disposições do acordo TRIPS que fornecem a flexibilidade necessária a esse propósito.
A licença compulsória nos casos de interesse público, e desde que o titular da patente não atenda a essa necessidade, também está prevista no art. 71 da Lei nº 9.279/96. Com base nele, inclusive, o TCU, no Processo 009.253/2015-7, recomendou ao Ministério da Saúde que avaliasse a possibilidade e a conveniência de concessão de licenças compulsórias para exploração de patentes de medicamentos a serem eventualmente incorporados ao SUS.
É certo que a licença compulsória é medida extrema que deve ser empregada com cautela e somente como último mecanismo de viabilização de acesso a um tratamento médico necessário aos cuidados adequados da saúde pública. Deve-se priorizar, portanto, a negociação de preços com o titular da patente e a licença voluntária. Ainda assim, a quebra de patente não pode ser descartada ou tomada como tabu. Foi com ela, aliás, que o Brasil serviu de exemplo para o mundo no tratamento do vírus da HIV pelo SUS.
9 Conclusão
Há um claro descompasso – não apenas no Brasil, mas no mundo todo – entre os preços dos medicamentos, em especial das tecnologias novas e inovadoras que chegam ao mercado, e a capacidade econômica dos consumidores, dos sistemas públicos e privados de saúde.
A correlação de forças entre os interesses econômicos da indústria farmacêutica e os interesses dos Estados e dos cidadãos nos cuidados com a saúde pública está claramente pendendo para os primeiros.
Há, no entanto, diversas determinantes comerciais positivas que, se corretamente empregadas, podem reequilibrar essa relação, permitindo que o ganho de uma das partes não se dê às custas da perda da outra. Privilegiar a acessibilidade aos medicamentos não significa desestimular a atividade econômica ou o investimento privado em pesquisa e desenvolvimento. Significa compatibilizar os diversos interesses, sem que um implique a aniquilação do outro.
Para tanto, várias medidas foram sugeridas no decorrer deste estudo, quais sejam:
- maior transparência da indústria farmacêutica em relação aos custos com pesquisa e desenvolvimento de tecnologias de saúde, composição e negociação de preços dos medicamentos e margem de lucro na sua comercialização;
- aprimoramento dos critérios de precificação dos medicamentos e de reajuste das tabelas da CMED;
- incentivo à celebração de acordos de compartilhamento de riscos e de pagamentos baseados em desfecho;
- revisão das estratégias de investimento em pesquisa e desenvolvimento de novas tecnologias em saúde; e,
- compatibilização entre os direitos de propriedade intelectual – patentes – e a acessibilidade econômica de medicamentos à sociedade, inclusive, quando necessário, mediante licenciamento compulsório.
Nenhuma dessas medidas, isoladamente, será a panaceia para o problema. No entanto, todas juntas, utilizadas de forma coordenada e com critérios técnicos qualificados, certamente contribuirão muito para que os medicamentos sirvam ao principal propósito a que se destinam, que é cuidar da saúde. Para isso, eles precisam chegar aos pacientes.
Imagem foto de Etactics Inc na Unsplash